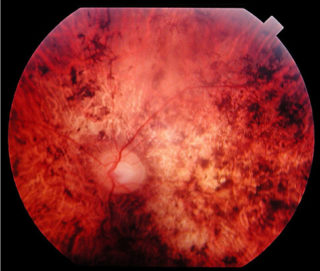
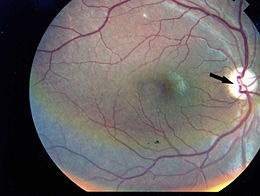
Il glioma del nervo ottico (astrocitoma di basso grado) è un tumore che appare sulla struttura che trasporta l’input visivo dall’occhio al cervello (astrociti del nervo ottico).
Generalmente il glioma nelle sue forme specialmente se riscontrate nei bambini piccoli, à benigno e facilmente curabile. Alcuni tumori negli adulti, tuttavia, evolvono rapidamente in cancerosi, crescendo in modo aggressivo e diffondendosi a altre parti del cervello e anche ad altri organi.
Il glioma del nervo ottico in entrambe le sue forme può provocare la perdita parziale o totale della vista, nonché mal di testa e contrazioni oculari.

STUDI SCIENTIFICI e CONNESSIONI GENETICHE
Ancora oggi non si comprende pienamente il motivo per cui questi gliomi del nervo ottico compaiano. Alcuni studi suggeriscono che la genetica giochi un ruolo nel loro sviluppo, poiché molti pazienti hanno storie familiari di tumori del tessuto nervoso.
Glioma e Neurofibromatosi di tipo 1
La prestigiosa rivista Cancers ha recentemente pubblicato uno studio relativo alle più moderne conoscenze sulle principali problematiche correlate al glioma del nervo ottico correlato alla Neurofibromatosi di tipo 1.
Tale articolo che ha visto coinvolti i diversi team di ricerca italiani e stranieri; partendo da un riesame della letteratura sull’argomento, sono state esaminate le attuali conoscenze relative al glioma delle vie ottiche evidenziando che questo tumore del sistema nervoso centrale colpisce fino al 20% dei pazienti affetti da neurofibromatosi. Vengono prese in esame la patogenesi, le attuali metodiche diagnostiche, la prognosi e le possibilità di trattamento di questi tumori, che possono provocare una riduzione anche molto importante della vista come anche diversi disturbi neurologici.
Fondamentalmente sono state esaminate le correlazioni genetiche che potranno un giorno consentire di prevedere quali pazienti siano a maggiore rischio di sviluppare tale neoplasia; ciò può portare alla messa a punto di protocolli di screening che permettano l’identificazione precoce del tumore anche in quei pazienti che non presentano sintomi. In questi casi, l’impiego della risonanza magnetica per la caratterizzazione delle lesioni a carico del sistema nervoso centrale e gli esami svolti dall’oculista specialista hanno un ruolo determinante nella diagnosi precoce e nel follow-up in caso di progressione della malattia.
Per quanto riguarda la terapia, accanto alla chemioterapia già in uso, si pone l’accento su alcuni nuovi promettenti farmaci, tuttora in fase di sperimentazione, capaci di interferire con i segnali biologici responsabili della crescita di questi tumori, che nelle prime fasi di sviluppo farmacologico hanno mostrato risultati promettenti, in particolare alcuni specifici inibitori di MEK-1/2.
Il lavoro citato ha analizzato il glioma del nervo ottico da un’ampia prospettiva multidisciplinare, che ha coinvolto numerose figure professionali dedicate alla cura del paziente e alla ricerca in tale ambito, e che mira ad una più completa comprensione dell’ originr e del comportamento di questi tumori come punto di partenza per lo sviluppo di nuove terapie con la speranza che possano avere un impatto significativo sulla vita dei pazienti.
SINTOMATOLOGIA
La maggior parte di questi tumori di natura benigna cresce molto lentamente e potrebbe non causare alcun sintomo fisico per diversi mesi o anni. È possibile che un bambino abbia gliomi su ciascun occhio, sebbene la maggior parte dei pazienti abbia problemi singolari. Le escrescenze maligne invece tendono a svilupparsi rapidamente, diventando cancerose e iniziando a diffondersi entro pochi mesi dal loro inizio.
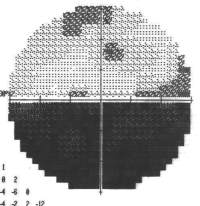
È probabile che un individuo che soffre di un glioma del nervo ottico si renda conto di un certo grado di compromissione della vista con diminuzione importante del visus. La perdita della vista periferica è più comune, ma un glioma avanzato può interessare tutti gli aspetti della vista. A seconda della pressione esercitata dal tumore sul nervo ottico, l’occhio interessato può sporgere verso l’esterno o contrarsi in modo incontrollabile. Alcuni pazienti manifestano sintomi generalizzati di affaticamento, mal di testa, nausea e deterioramento cognitivo.
Quando si sospetta un glioma del nervo ottico, il paziente deve essere indirizzato da un neurologo che può eseguire scansioni di tomografia computerizzata (TC) e test di risonanza magnetica (MRI) sugli occhi e sul cervello, alla ricerca di segni di masse insolite e cicatrici.
Una volta scoperto un tumore, si può scegliere di estrarre un piccolo campione di tessuto per analisi di laboratorio per rivelare se la massa è cancerosa, benigna o segno di un’altra patologia cerebrale più grave.
TRATTAMENTO
I tumori piccoli e benigni possono spesso essere rimossi chirurgicamente, anche se potrebbe essere necessario trattare una massa cancerosa con una combinazione di chemioterapia e radiazioni.
La chirurgia di solito può essere eseguita su un piccolo glioma del nervo ottico per asportare la massa e alleviare la pressione sul nervo. Se l’intervento chirurgico non ha successo o il cancro ha già iniziato a diffondersi, un paziente potrebbe dover sottoporsi a diversi cicli di radioterapia o chemioterapia.
Ai pazienti vengono in genere prescritti farmaci antidolorifici e viene chiesto loro di riposare gli occhi il più possibile durante il recupero. Il trattamento per i tumori benigni spesso porta al completo recupero della vista, anche se è probabile che i problemi maligni si traducano in una perdita permanente della vista.




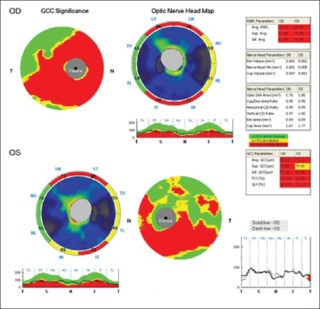
 delle stesse che può portare a gravi disturbi delle sinapsi fino alla morte delle cellule interessate.
delle stesse che può portare a gravi disturbi delle sinapsi fino alla morte delle cellule interessate.