Malattie genetiche rare
LE RETINOPATIE LEGATE AL CROMOSOMA X
Le retinopatie legate al cromosoma X rappresentano un gruppo di malattie ereditarie della retina, che costituiscono un’importante causa di cecità soprattutto nei bambini.
Trattandosi di un gruppo eterogeneo di malattie, anche i meccanismi patogenetici alla base di queste patologie possono essere differenti tra loro. Tuttavia, le retinopatie legate al cromosoma X sono solitamente causate da mutazioni genetiche che provocano una perdita della funzione di alcune proteine e, per questo, rappresentano un ottimo target per le strategie di terapia genica.
Molte delle retinopatie legate al cromosoma X vengono trasmesse alla progenie da madri portatrici sane (cioè che non sviluppano la malattia, ma possono trasmetterla) e questo, nel tempo, ha aiutato i medici a conoscere sempre meglio le modalità di trasmissione ereditaria di queste patologie.
Dati pubblicati di recente dal Center for Hereditary Retinal Degenerations della University of Pennsylvania hanno riscontrato una base genetica nel 52% dei pazienti con retinopatia e, di questi, il 17% aveva una retinopatia legata al cromosoma X.
In primo luogo la RETINITE PIGMENTOSA
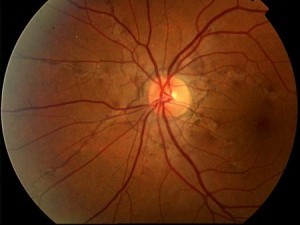
La retinite pigmentosa è una degenerazione lentamente progressiva e bilaterale della retina e dell’epitelio pigmentato retinico, causata da varie mutazioni genetiche. I sintomi comprendono emeralopia e riduzione del campo visivo periferico. La diagnosi si basa sull’esame del fondo oculare, che mostra pigmentazione a forma di spicole ossee nella retina equatoriale, restringimento delle arteriole retiniche, pallore cereo del disco ottico, cataratta sottocapsulare posteriore e cellule nel vitreo. L’elettro-retinogramma è utile per confermare la diagnosi. La vitamina A palmitato, acidi grassi omega-3 e luteina più zeaxantina possono contribuire a rallentare la progressione della perdita della vista.
La retinite pigmentosa sembra essere causata da un gene anomalo che codifica per le proteine retiniche; sono stati identificati parecchi geni. La trasmissione può essere autosomica recessiva, autosomica dominante o, raramente, legata al cromosoma X. Può rientrare nell’ambito di una sindrome (p. es., bassen-Kornzweig, Laurence-Moon). Una di queste sindromi comprende pure la perdita congenita dell’udito (sindrome di Usher).
L’ereditarietà legata al cromosoma X e le modalità genetiche di trasmissione delle retinopatie
Le malattie causate da variazioni di sequenza nei geni presenti sul cromosoma X sono note come malattie “X-linked”.
I cromosomi X e Y sono detti cromosomi sessuali, perché determinano il sesso del nascituro (XX per le donne e XY per gli uomini). Il cromosoma X contiene circa 1000 geni, rispetto ai circa 70 presenti sul cromosoma Y. Per equilibrare questa grande differenza, uno dei due cromosomi X, nelle donne, va incontro a un fenomeno detto “inattivazione”. Si tratta di un processo fisiologico che causa il silenziamento casuale di uno dei due cromosomi X, i cui geni, di conseguenza, non vengono espressi.
È stato dimostrato che alcuni dei geni legati al cromosoma X, che sono stati individuati tra le cause di malattie retiniche, come i geni RPGR, RP2 e CACNA1F (associati alla retinite pigmentosa e ad altre retinopatie) subiscono tutti una completa inattivazione.
Alcuni geni legati allo sviluppo di retinopatie
Come detto, le forme di retinopatie legate a mutazioni o inattivazione dei geni presenti sul cromosoma X sono molteplici. Tra queste, alcune delle più frequenti, sono legate ai seguenti geni:
RPGR (retinitis pigmentosa GTPase regulator) è stato il primo gene identificato come causa della retinite pigmentosa legata al cromosoma X.
Le mutazioni di RPGR sono responsabili di diversi tipi di malattia, tra cui la distrofia dei bastoncelli-coni (70%), la distrofia dei coni-bastoncelli (6-23%) e la distrofia dei coni (7%). (degenerazione primaria dei bastoncelli/coni, associata ad un marcato interessamento secondario dei coni/bastoncelli, con aspetto variabile del fondo oculare)
La retinite pigmentosa (RP) mostra un’ereditarietà legata al cromosoma X nell’8-16% dei pazienti con una prevalenza di maschi affetti di circa 1:15.000-1:26.000.
Il gene RPGR è responsabile di oltre il 70% di questi casi. La RP legata al cromosoma X tende ad avere un fenotipo più grave e si presenta spesso durante l’infanzia (in media a 5 anni di età).
Il gene della retinite pigmentosa RP2 è stato il secondo gene identificato come causa di RP legata al cromosoma X. Le mutazioni di questo gene sono responsabili di circa il 10-20% dei casi di RP legata al cromosoma X. I pazienti presentano caratteristiche tipiche della RP, tra cui cecità notturna, costrizione del campo visivo e conseguente riduzione dell’acuità visiva.
Varianti del gene CHM sono responsabili della degenerazione corioretinica nella coroideremia (degenerazione progressiva dell’epitelio pigmentato retinico (EPR) e della coroide con prognosi sfavorevole per la vista nella quasi totalita’ dei casi).
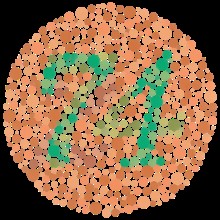
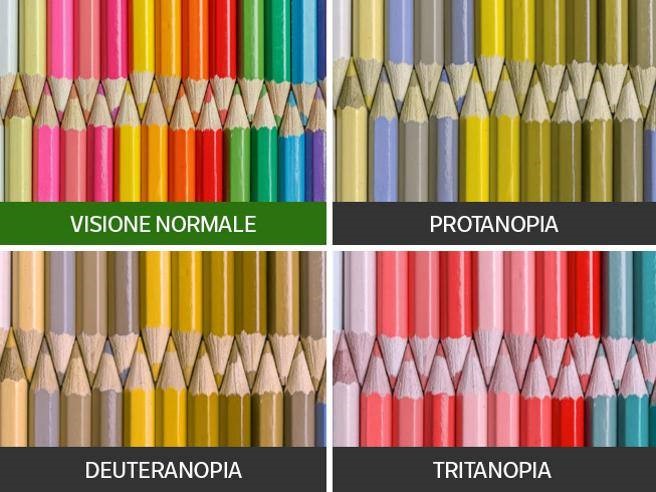
La coroideremia colpisce da 1:50.000 a 1:100.000 persone, con un’alta prevalenza in Finlandia. I maschi sviluppano sintomi di nictalopia nella prima decade di vita, seguiti da una costrizione del campo visivo progressiva. È stato anche riportato che i pazienti con coroideremia hanno una riduzione generalizzata della visione dei colori, evidente già all’inizio della malattia.
La coroideremia è solitamente una malattia retinica isolata, tuttavia sono state osservate anche associazioni sindromiche. In questi casi la patologia è associata a perdita dell’udito, deterioramento cognitivo, labbro leporino e palatoschisi, deformità scheletriche, acrocheratosi.
La retinoschisi legata al cromosoma X è una condizione che colpisce approssimativamente 1:15000-1:30.000 individui maschi, che tipicamente presentano sintomi di riduzione della visione centrale in età scolare, strabismo o anisometropia. La prognosi è spesso relativamente buona nell’infanzia, a meno che non si verifichino distacco di retina o emorragia del vitreo, che sono associati a una prognosi infausta. Circa il 50% dei pazienti presenta anche alterazioni retiniche periferiche. Un sottogruppo di pazienti presenta una retinoschisi bollosa, che tende a presentarsi nell’infanzia con strabismo, riduzione significativa della vista, nistagmo, mosche volanti secondarie a emorragia del vitreo o pupilla di forma irregolare.
Terapie geniche sperimentali
Attualmente per le retinopatie sono in fase di sperimentazione clinica una serie di nuove terapie, che probabilmente verranno ulteriormente sviluppate nei prossimi anni. Gli approcci terapeutici possono essere classificati in due tipi: quelli che mirano a rallentare il tasso di degenerazione e ridurre al minimo la perdita della funzione visiva e quelli che puntano a ripristinare la funzione visiva nella malattia allo stadio terminale.
La terapia genica è l’approccio terapeutico più avanzato per le varianti che causano una perdita di funzione ed è già disponibile un trattamento di sostituzione genica autorizzato per la malattia autosomica recessiva associata al gene RPE65.
Altri metodi sono impiegati in diverse fasi di sviluppo e non sono stati ancora applicati alle retinopatie legate al cromosoma X, ma sembrano promettenti. Questi includono gli oligonucleotidi antisenso (che sono piccole molecole che alterano l’espressione dell’RNA), l’editing genetico (ad esempio la tecnica di recente sviluppo CRISPR/cas-9) e l’editing dell’RNA.
Altri approcci includono farmaci locali o sistemici, volti a migliorare la sopravvivenza cellulare.
Bibliografia
Samantha R.De Silva et al., The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies, Progress in Retinal and Eye Research


 Le probabilità che il nascituro sviluppi complicanze si riducono al 30%, se l’infezione avviene tra la 11esima e la 16esima settimana. Nelle infezioni contratte oltre la 17esima settimana di gravidanza, nel neonato è stato registrato prevalentemente un rischio di sordità congenita o problemi all’apparato visivo. Oltre il primo trimestre di gestazione, infatti, la placenta esplica un’azione protettiva, quindi è più raro che si verifichi un’infezione fetale in questo periodo.
Le probabilità che il nascituro sviluppi complicanze si riducono al 30%, se l’infezione avviene tra la 11esima e la 16esima settimana. Nelle infezioni contratte oltre la 17esima settimana di gravidanza, nel neonato è stato registrato prevalentemente un rischio di sordità congenita o problemi all’apparato visivo. Oltre il primo trimestre di gestazione, infatti, la placenta esplica un’azione protettiva, quindi è più raro che si verifichi un’infezione fetale in questo periodo.