Atrofia ottica dominante
L’atrofia ottica è trasmessa geneticamente e comporta l’atrofia delle cellule ganglionari della retina e di conseguenza porta all’atrofia del nervo ottico.
L’atrofia ottica dominante, o atrofia di Kjer, è una malattia genetica e così come la sindrome si Leber, è progressiva e porta in ambedue gli occhi all’atrofia del nervo ottico.
Nella patologia in questione l’età di esordio è precoce; dall’età prescolare ai 6/7 anni. Il bambino in poco tempo, a volte giorni o settimane, subisce un rapido calo del visus. Di solito i primi ad accorgersi del disagio sono gli insegnanti; spesso il disturbo viene scambiato per deficit di attenzione, dislessia o deficit visus-temporale. In realtà un’attenta anamnesi permette di orientarsi verso l’atrofia ottica dominante perché spesso in famiglia esistono, o sono esistiti, altri casi.
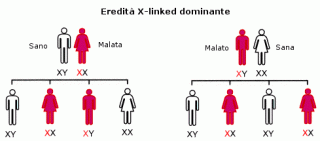
Quello mi sento spesso ripetere è “Avevamo uno zio (nipote, nonno) cieco”. In questa affezione esiste una mutazione del gene OPA1 che codifica una proteina mitocondriale responsabile dei fattori di respirazione cellulare.
Un genitore affetto ha una probabilità del 50% di trasmettere la malattia ai propri figli.
Diagnosi
Il bambino affetto arriva alla visita specialistica con i dischi ottici con un visus, spesso differente tra i due occhi, fra 2 e 6/10 anche se ci sono casi più drammatici in cui il visus; probabilmente per un ritardo verso la comprensione del problema, può essere anche molto più compromesso.
Al campo visivo si osserva uno scotoma centrale che spesso si allarga verso la macchia cieca (centro ciecale) o che coinvolge il campo visivo inferiore, come a simulare una sorta di emianopsia altitudinale (mancanza di 1/2 campo visivo in orizzontale).
Al test per la visione cromatica solitamente si evidenziano deficit per la distinzione dell’asse blu/giallo (tritanopia), ma anche di quello rosso/verde (protanopia).
Talvolta possono essere associati anche disturbi dell’udito.
Esami clinici
Il laboratorio di genetica stabilisce il deficit del gene OPA1 per avere la diagnosi definitiva della sidrome.
Altro esame importante è l’esame dei potenziali evocati visivi (PEV) da pattern; (si mette il bambino di fronte ad uno schermo con scacchi bianchi e neri in cui a scatti l’immagine viene invertita mentre degli elettrodi vengono posti nella regione occipitale al di sopra della nuca per monitorare il segnale elettrico che arriva alla corteccia visiva); i risultati nel caso di atrofia ottica sono nulli; (la variazione dell’immagine non viene rilevata) o nei casi di semi-atrofia si osserva una forte riduzione nell’ampiezza del segnale rilevato.
Un altro esame elettro-fisologico è ERG da pattern (o PERG) nel quale l’elettrodo a foglia dorata è appoggiato direttamente sulla cornea; il segnale rilevato è espressione dello strato retinico interno.
Questa forma ha un’incidenza di 1/50.000; quello che differenza la sindrome di Leber e l’atrofia ottica dominante è che la prima comporta una riduzione visiva in alcune settimane verso i 30 anni o poco prima, mentre la seconda compare in età infantile. Le donne affette da sindrome di Leber possono talvolta manifestare la malattia anche dopo i 40 anni.
Terapia
Un farmaco che può avere un’azione positiva è l’idebenone o raxone; inizialmente utilizzato nella sindrome di Alzheimer, nell’atassia di Frededich e nella distrofia di Duchenne dove non ha ottenuto risultati significativi.
L’azione di questo principio attivo, che agisce come antiossidante della cellula, è quella di trasportare elettroni per i mitocondri che producono l’ATP (Adenosine Tri-Phosfato) che è la sorgente di energia dei cicli organici della cellula e inibisce la formazione dell’ipeperossido che è un acido di scarto nocivo per la cellula.
Il problema principale dell’idebenone è che viene assorbito enormenente dal fegato; solo l’1% raggiunge la circolazione, per questo occorre usare dosaggi elevati e migliorarne l’assorbimento assumendolo con cibo grasso; personalmente utilizzo preparazioni galeniche magistrali che contengono olio di oliva.
Se vogliamo potenziare ulteriormete l’azione antiossidativa dobbiamo associare l’ubichinone o coenzima Q e l’acido lipoico.
Come terapia nella fase acuta, quando il nervo ottico si presenta con bordi edematosi, è consigliabile ricorrere a cortisonici; spesso questi riducono l’edema papillare.
In conclusione la terapia sopra riportata non ha chiaramente lo scopo di portare a guarigione malattie genetiche rare; l’obiettivo è quello di salvaguardare il più possibile le strutture nervose implicate nella speranza che la ricerca genetica approdi entro tempi ragionevoli all’applicazione clinica.





2 comments
Pingback: Atrofia ottica: nuove prospettive terapeutiche | Carmine Ciccarini
Pingback: Atrofie ottiche anomale: casi clinici | Carmine Ciccarini
Comments are closed.