FOCUS MALATTIE RARE CON SINTOMI OFTALMOLOGICI:
Questo articolo vuole illustrare due malattie rare di trasmissione genetica che presentano distinti sintomi oftalmologici: se non correttamente valutate possono essere confuse con altre patologie.

LA SINDROME DI KEARNS – SAYRE
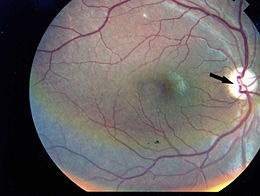
La sindrome di Kearns-Sayre (KSS) è una malattia rara (1/120.000 soggetti) generata da un raro errore congenito del metabolismo; essa è caratterizzato da oftalmoplegia esterna progressiva (PEO – con questo acronimo si intende una delle manifestazioni cliniche più comuni delle malattie mitocondriali; caratteristiche fondamentali di questa sindrome sono la ptosi palpebrale e la progressiva debolezza dei muscoli che governano i movimenti degli occhi) e retinite pigmentosa e per questo confusa appunto con la PEO progressiva.
La differenza tra le due patologie è che la prima esordisce in giovane età, la seconda invece colpisce soggetti adulti tra i 60 e gli 80 anni.
Entrambe le sindromi causano ptosi palpebrale (palpebra cadente) con paralisi dei muscoli oculari, con sguardo del paziente fisso e impossibilità di movimento degli occhi.
Altri sintomi associati spesso alla sindrome sono la presenza di retinite pigmentosa e diversi sintomi di natura neurologica: difficoltà nella parola, deficit cognitivo, cardiopatia, sordità e atassia cerebellare, non necessariamente presenti insieme.
La trasmissione genetica della sindrome di Kearns-Sayre avviene per via materna essendo trasmessa dalla cellula uovo; si tratta di una malattia mitocondriale, colpiscono cioè il mitocondrio che è il fornitore dell’energia del neurone. I geni coinvolti nella mutazione sono multipli e rappresentano la causa di anomalie multiple del DNA mitocondriale. La trasmissione può essere sia dominante che recessiva.
DIAGNOSI
La diagnosi genetica prevede il sequeziamento (tramite materiale raccolto con biopsia muscolare) del DNA mitocondriale e dei geni nucleari che permette di diagnosticare la mutazione.
Esistono comunque varianti della sindrome (es. sindrome di Melas e sindrome di Merrf) che associano altri sintomi come perdita totale di vista e udito e forme di epilessia.
TERAPIA
Le cure per la sindrome di Kearns-Savre sono essenzialmente sintomatiche.
Buoni risultati si possono ottenere somministrando Idebenone, per salvaguardare la vita delle cellule mitocondriali, in associazione a zafferano ad alti dosaggi che riduce gli eventi infiammatori che sono alla base di tutte le patologie degenerative.
Al momento non esistono terapie geniche, ma sono in corso studi a questo proposito in molti laboratori di ricerca.
LA SINDROME KIF2IA
Sindrome genetica estremamente rara che comporta fibrosi congenita dei muscoli extra-oculari. Fin dalla nascita i neonati presentano subito una ptosi palpebrale e oftalmoplegia (alterazione della motilità oculare). I bambini non sembrano più in grado di gestire i muscoli interessati tanto che talvolta presentano atteggiamenti di esotropia o exotropia con impossibilità nei movimenti oculari.
Questa mutazione risulta molto più diffusa nelle popolazioni di origine libanese, siriana o marocchina; i rarissimi casi che si osservano in Italia sono quasi sempre in bambini con genitori naturali di questa provenienza (immigrati o adottati).
Per gli effetti legati alla grave ptosi i piccoli pazienti si presentano con il mento sollevato (effetto chin-up), ma per il resto la visione risulta assolutamente normale lo stesso si evidenzia sia per le funzioni cognitive che motorie.
La sindrome presente nel cromosoma K causa un danno della kinesina, una proteina necessaria ad un perfetto funzionamento neuro-vascolare. La malattia è trasmessa in eterozigosi, ma anche in forma dominante; esistono anche casi di ambliopia, coloboma del nervo ottico, glaucoma e cataratta.
TERAPIA
La terapia è di natura esclusivamente chirurgica e consiste nell’intervento sulla ptosi palpebrale congenita.
Alcuni pensano che intervenendo con neurochirurgia o chirurgia maxillo-facciale sulla fibrosi dei muscoli retrobulbari si possa avere un miglioramento nella motilità oculare, ma al momento nessuno ha mai effettuato questo trattamento sperimentale.
malattia di Vogt-Koyanagi-Harada
La malattia di Vogt-Koyanagi-Harada è una patologia sistemica molto rara, l’incidenza è stimata in circa 1/400.000 casi l’anno, le donne tra i 20 e i 30 anni sono colpite di più degli uomini
La sindrome si manifesta con una panuveite granulomatosa diffusa, cronica, bilaterale, caratterizzata dal distacco della retina di tipo essudativo, spesso associata a alterazioni neurologiche (meningite), dell’udito e della cute.
È particolarmente diffusa tra i soggetti di origine asiatica, indiana, nei discendenti degli indiani d’America e negli Ispanici, ad essa è infatti riconosciuta una base genetica che si ipotizza sia legata all’alterazione di alcune proteine motrici denominate chinesine; tale alterazione genera una risposta autoimmune contro normali costituenti dell’organismo, nello specifico contro i melanociti, cellule contenenti melanina, localizzate nel tratto uveale, nella cute, nell’orecchio interno e nelle meningi.
SINTOMATOLOGIA
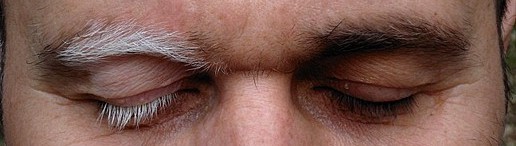
L’esordio della malattia è di solito sistemico ed aspecifico: febbre, cefalea, indolenzimento, ma subito dopo sintomi neurologici tendono a manifestarsi precocemente e comprendono acufeni, abbassamento dell’udito, vertigini, cefalea e meningismo. I riscontri cutanei compaiono spesso più tardi e comprendono vitiligine a chiazze (particolarmente frequente su palpebre, regione lombare e natiche), poliosi (una chiazza localizzata di capelli bianchi, che può coinvolgere le ciglia) e alopecia.
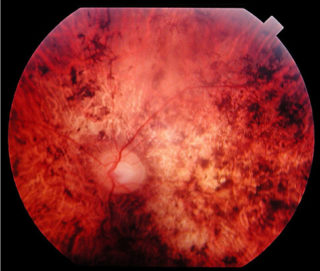
La fase oftalmologica compare pochi giorni dopo; il paziente lamenta visione offuscata, dolore ai globi oculari e scotoma centrale (bilaterale nell’80% dei casi). Spesso si verifica un distacco della retina, bilaterale, di tipo essudativo, edema del disco ottico e coroidite.
Le uveiti ricorrenti caratterizzano l’ultimo stadio, la fase cronica. Altre complicanze sono la comparsa di cataratta, glaucoma, fibrosi sottoretinica, e neo-vascolarizzazione coroideale.
TRATTAMENTO
Il trattamento precoce comprende corticosteroidi locali e sistemici e un farmaco cicloplegico-midriatico (che agisce dilatando la pupilla). Molti pazienti rispondono inoltre al trattamento con immunosoppressori non-steroidei.
Utilizzando un trattamento precoce ed aggressivo la prognosi di solito è favorevole, anche se possono recidivare i disturbi acuti alla vista e all’udito.